
Chapter 7 - Conducting Your Study
Consent Process
Key points
- Investigators will not enroll or involve a subject in any research activities, until legally effective informed consent has been obtained unless need for consent has been waived by the IRB.
- The informed consent document must be signed and dated by the subject, parent or the LAR, and a copy of the signed informed consent document must be provided to the subject who signed it (except when the IRB has approved a waiver of documentation of informed consent).
- For NIH investigators, the consent process must be documented in the subject's record and include a description of the method used for communication with the subject and the specific means by which the subject communicated agreement to participate in the study (e.g., their verbal response and signing of the informed consent document).
- When research involves children, no child may be enrolled, screened, or have research procedures initiated, unless parental permission and child assent is obtained unless waived by the IRB.
- When obtaining consent or assent from a participant who is considered part of a vulnerable population or who does not understand English, the applicable subparts (B-D) of the federal regulations for the protection of human subjects and NIH HRPP policies must be followed.
- Additional information can be found in NIH Policy 301, Informed Consent.
What do I need to know about the consent process?
- Be sure to use the most recent IRB-approved version of the consent form. The consent should have a stamp with the IRB approval date. The most current version will be located at this website or in the electronic IRB system.
- Whenever possible, you should provide the potential subject with the consent form before the consent discussion so that they have time to read it and discuss with others if they desire (e.g., family member, their primary care provider).
- Subjects need to be provided with sufficient opportunity to read the consent form and discuss it with a knowledgeable investigator on the protocol before deciding whether to participate.
- The consent discussion should be conducted in a way that minimizes possibility of coercion or undue influence.
- The consent process should be conducted in a private, quiet location to the greatest extent possible.
- Subjects can sign the consent form either in writing or electronically. However, when obtaining electronic signature methods for obtaining electronic signature must comply with any NIH or IC-specific and IRBO requirements. In addition, the process for obtaining electronic signature must be prospectively reviewed and approved by the IRB.
- The research subject is given a copy of the signed informed consent document.
- At the Clinical Center, the original signed informed consent document must be forwarded to the Health Information Management Department for review and inclusion in CRIS.
- In cases in which the documentation requirement is waived, the IRB may require the investigator to provide participants or the parents of children who are participants with a written copy of the consent information or a statement regarding the research.
- The consent process must be documented in the consent note in the subject's CRIS/medical record (or the research record if there is no medical record) regardless of whether documentation of consent was waived or not.
What is the process for obtaining remote consent by phone or virtual platform?
- Remote consent processes can be used but must be described in the protocol and approved by the IRB prior to use.
- The subject should be provided with the consent form in advance of the consent conversation.
- After the consent process has been conducted and the investigator has responded to the subject's questions, the subject signs the consent form noting the current date.
- The investigator documents the process in the consent note in CRIS/medical/research record in real time on the day of the consent conversation.
- When the signed/dated consent form is returned to the investigator who conducted the consent discussion, the investigator signs and dates the consent form with the date they received the signed the consent from the subject.
- The investigator should then record another note in CRIS/medical/research record indicating the updated status, send a copy to medical records (or research record if there is no medical record), and provide a copy of the completed consent form to the subject.
- The date that the subject signs the consent form is considered their "date on study."
- If, after the subject has signed the consent form, specimens and/or data are collected locally for research purposes, no analyses of these specimens and/or data may occur until the investigator has verified that the subject has returned a signed and dated informed consent document, unless the IRB has granted a waiver of documentation of consent.
For enrollment of subjects who do not read English, what is the difference between using a translated long form consent and using the short form consent process?
- The translated long form consent can be used when it is written in the language that the potential subject can read.
- When non-English speaking subjects are anticipated to enroll in the research, you must submit a certified translated long form consent document in the language of the anticipated subjects to the IRB and receive IRB approval before the translated long form consent document is used.
- When non-English speaking subjects are anticipated to enroll in the research, you must submit a certified translated long form consent document in the language of the anticipated subjects to the IRB and receive IRB approval before the translated long form consent document is used.
- The short form consent process is used when the subject is unable to read the long form version of the consent due to a language barrier. An interpreter is utilized for subjects who are unable to understand the language in which the long form consent is written. The IRB must approve the document that will be used as the basis for the consent conversation, and this is usually the English long form consent. See below for additional information on use of the short form consent process.
What else do I need to know about using a short form consent process?
- See FAQs Everything you need to know about consent for more information about the consent process for enrolling subjects who do not speak English.
- If enrollment of a non-English speaking subject is not anticipated, an IRB-approved short form consent in the language of the subject must be used. Translated short form consents are available on the OHSRP website on the Short Form Consents webpage
- If there is no IRB-approved short form consent document in the language of the subject, the NIH PI must submit to the IRB for approval (and before use), a certified translation of the short form consent in the language of the subject.
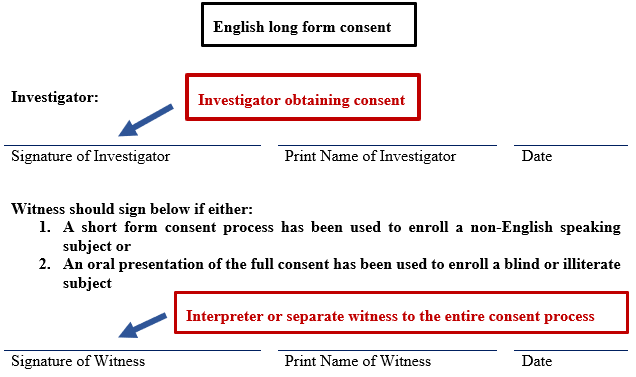
- An individual serving as the witness must be present at the site of the investigator obtaining consent and must observe the entire consent discussion.
- The witness must be fluent in the language of the subject and in English.
- In the vary rare instance that the translator is unable to act as the witness, and if the witness is not fluent in both the language of the subject and English, then the witness should verify the following with the interpreter:
- The subject understands the information presented.
- All questions have been satisfactorily addressed.
and - The subject agrees to participate.
- The witness, or investigator obtaining informed consent, should document the short form consent procedure in the consent note in CRIS/medical/research record.
- When the short form process is used:
- The investigator who obtains consent signs the English long form consent used as the basis of the consent discussion.
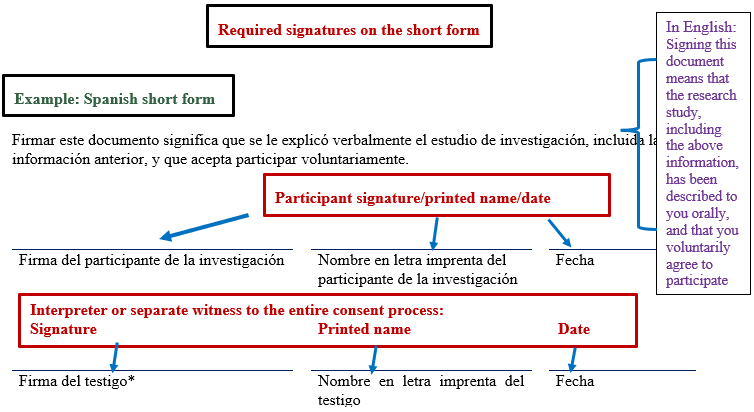
- The witness (who may be the interpreter though a separate witness will be needed if the interpreter cannot/does not wish to act as the witness) signs the English long form consent and the short form consent.
- The participant signs the short form that is in the language that they can read.
________________________________________________________________________________________
- If the investigator is truly fluent in English and the language of the subject, and there is no IRB approved translated long form, the short form process should be used as follows:
- The bilingual investigator conducts the consent process in the language of the participant and explains all applicable elements of consent using the English long form as the summary of what is said to the participant.
- The investigator obtaining consent cannot act as the witness, so the second option in the administrative block is checked, and the investigator's name is noted on the provided line.
- In such cases, there must be a separate bilingual individual present to observe the entire consent process who signs as the witness.

- Additional information regarding requirements for the short form consent process are included in Policy 301 Informed Consent.
How is the consent process conducted when a translated long form is used?
- If investigators anticipate enrolling participants who speak a specific language other than English, the English long form consent should be fully translated into that language and submitted to the IRB for approval prior to use.
In this case, an interpreter is generally also used to facilitate the discussion and answer the participant's questions.
1- The investigator obtaining consent and the participant both sign the fully translated long form consent (as they would if the long form was in English), and a witness is not required on the long form.
- The second box in the administrative section should be checked to indicate that the interpreter facilitated the consent process but did not serve as the witness. (See screenshot below.)

What do I do when the potential subject speaks and understands English but is blind or illiterate?
- When the subject speaks and understands English but is illiterate or blind, the English long form should be used to obtain consent from the subject. The short form consent document should not be used.
- The subject may use assistive technology to read the consent (such as screen readers for sight-impaired individuals), or the consent form should be read to the subject.
- There must be a witness to the entire oral presentation of the consent. The witness then signs the witness line on the English long form consent. Subjects who are unable to sign their name can make their mark on the signature line. (e.g., They may make an "X," or provide a fingerprint.)
- The consent note in CRIS/medical/research record should document the process and include a statement that there was a witness to the entire consent process and any special circumstances regarding documentation of consent.

What should I do when conducting consent with a potential subject who does not speak English and for whom there is no written language?
- When conducting the consent process with a subject for whom no written language exists, the process is similar to that used with a blind or illiterate subject.
- There should be an oral presentation of the English long-form consent by the interpreter.
- There must be a witness (who can be the interpreter if they are willing to act as the witness) at the location of the investigator, who is present during the entire oral presentation.
- The subject must sign or make their mark on the consent, and the investigator and witness both sign the consent.
- The administrative block for interpreters must be completed, and the consent process used in this circumstance must be documented in the consent note in CRIS/medical/research record.

Consent process when participants are considered a vulnerable population
Key points
- Federal regulations for the protection of human subjects participating in research as well as NIH policy include specific requirements when enrolling subjects from special populations.
- Investigators are expected to understand and utilize the required process if they are enrolling subjects from these groups.
What other considerations about the consent process exist when doing research with pregnant persons?
- In this case, it is the fetus that is considered vulnerable and not the pregnant woman.
- If the IRB approves the enrollment of pregnant women on the protocol, the consent of the woman is sufficient UNLESS the research holds out the prospect of direct benefit solely to the fetus in which case the consent of the pregnant woman and the father is required (unless the father is unable to consent because of unavailability, incompetence, or temporary incapacity or the pregnancy resulted from rape or incest).
- Pregnant women/fetuses or in vitro fertilization cannot be involved in emergency research where the requirement for informed consent has been waived by the Secretary under the authority of 45 CFR 46.101.
- Specific regulations apply if research will include neonates. In such cases, see section §46.205 Research involving neonates or contact the IRBO for additional questions.
See this section of the investigator manual which explains the conditions under which the IRB can approve research conducted with pregnant women.
What other considerations about the consent process exist when doing research with prisoners?
- Informed consent can be waived or altered in research involving prisoners so long as the appropriately constituted IRB reviews the research and makes the appropriate findings regarding the waiver or alteration of informed consent requirements. However, even if informed consent is waived or altered, federal regulations still require that the subjects be clearly informed in advance that participation in the research will have no effect on their parole, if such notification is relevant.
- The IRB will usually require separate consent documents be used if the subject being enrolled is a prisoner.
- Prisoners cannot be involved in emergency research where the requirement for informed consent has been waived by the Secretary under the authority of 45 CFR 46.101.
There are specific regulatory criteria that must be met prior to enrollment of prisoners in research or if an existing subject becomes incarcerated while on study. See this section of the manual for information on research with prisoners.
What other considerations about the consent process exist when doing research with children?
- When research involves children, no child may be enrolled, screened, or have research procedures initiated, unless parental permission and child assent is obtained unless waived by the IRB.
- The IRB may decide that the permission of one parent is sufficient in some cases (i.e., research does not involve greater than minimal risk or the research involves greater than minimal risk but presents prospect of direct benefit to the individual minor subjects). However, for research taking place at an NIH site, in cases where parents share joint legal custody for medical decision-making of a child (e.g., by a custody agreement or court order), both parents must give their permission regardless of the risk level of the research. Exceptions may include if one parent has since died, become incompetent, or is not reasonably available.
What does it mean to obtain a child's assent to participate in my research study?
- Assent is a regulatory requirement and means the affirmative agreement to participate in research. Mere failure to object should not, absent affirmative agreement, be construed as assent. This means the child must actively show his or her willingness to participate in the research, rather than just complying with directions to participate and not resisting in any way.
- When judging whether children are capable of assent, the IRB is charged with taking into account the ages, maturity, and psychological state of the children involved. The IRB has the discretion to judge children's capacity to assent for all of the children to be involved in a proposed research activity, or on an individual basis.
- The IRB may waive assent of the child only when specific conditions are met:
- The IRB determines that the capability of some or all of the children is so limited that they cannot reasonably be consulted (e.g., limited due to very young age or cognitive capacity of the potential minor participants).
OR - The intervention or procedure involved in the research holds out a prospect of direct benefit that is important to the health or well-being of the children and is available only in the context of the research.
- The IRB determines that the capability of some or all of the children is so limited that they cannot reasonably be consulted (e.g., limited due to very young age or cognitive capacity of the potential minor participants).
- The OHSRP website has information about the assent process and has a recommended assent template (at the bottom of that same information page). Children who are at least 7 years old who do not have underlying cognitive impairment are usually considered to have capacity to assent to participation in research.
How should assent by the minor subject for research participation be documented?
- The HHS regulations do not require documentation of assent. The IRB has the discretion to determine the appropriate manner, if any, of documenting child assent. Based on considerations such as the child's age, maturity, and degree of literacy, the IRB should decide what form of documentation, if any, is most appropriate.
- If young children who are unable to read are involved, documentation should take a form that is appropriate for the purpose of recording that assent took place. The IRB may also decide that documentation of assent is not warranted.
- When the IRB determines that assent is required, it determines whether and how assent must be documented. The assent process may be either oral or written.
- If you wish to have the IRB waive assent for some or all of the participants in your study, this should be described in the protocol and should include the conditions under which the waiver will apply. In the absence of a waiver, you must obtain assent. As noted above, the IRB can waive this requirement based on a number of factors such as the age of the children who will be participating as well as the cognitive capacity of potential participants.
- While assent can be written or verbal, the NIH IRB prefers that investigators submit an age-appropriate written assent form which should be written in a simple, readable style based on the intended minor subjects.
- Since the regular consent document should be written as close to an 8th grade reading level as possible, you may consider having older children (ages 14 and up) sign the regular consent as their assent document, as they are able to read and understand that document. (See screenshot below.) Plans to utilize such a process for older minors needs to be described in the protocol and approved by the IRB.

What is the process regarding consent when minor participants on my study reach adulthood?
- When children reach the age of majority under the applicable law of the jurisdiction in which the research will be conducted (18 years of age at the CC), they must provide consent in order to continue participation in the research, unless consent is not required (e.g., for certain exempt research) or, alternatively, a waiver of consent must be approved by the IRB.
- If the child who has reached the age of majority lacks the capacity to consent to the research, the NIH investigator will follow the requirements summarized below and in Policy 403 Research Involving Adults Who Lack Decision-making Capacity to Consent to Research Participation
unless the IRB has waived consent.
- This requirement applies not only when you will continue to interact directly with the now-adult subject but also whenever the protocol continues to meet the regulatory definition of "human subjects research." For example, if your protocol involves only the continued analysis of specimens and/or data for which the subject's identity is readily identifiable to you, then you need to seek and obtain the legally effective informed consent of the now-adult subject unless the IRB approves a waiver of consent.
For additional information on conducting research with children, see the section of this manual that relates to research with children.
Can parental or guardian permission for research involving children be waived?
- Under certain circumstances, an IRB may waive the requirements for obtaining parental or guardian permission if it makes and documents specific findings.
- In addition to the provisions for waiver contained in the section of the regulations that relates to requirements for informed consent, if the IRB determines that a research protocol is designed to study conditions in children or a subject population for which parental or guardian permission is not a reasonable requirement to protect the subjects (for example, neglected or abused children), it may waive the parental permission requirements provided that an appropriate mechanism is in place to protect the children, and provided that the waiver is not inconsistent with federal, state, or local law. The choice of an appropriate substitute mechanism (for example, appointing a child advocate or an assent monitor) for protecting children participating in research would depend on the nature and purpose of the activities described in the protocol, the risk and anticipated benefit to the research subjects, and the child's age, maturity, status, and condition (45 CFR 46.408(c)).
What considerations about the consent process exist when doing research with adults who lack capacity to consent to research participation?
- If you plan to enroll subjects who lack the capacity to consent to research participation, this must be described in the protocol and prospectively approved by the IRB. If this is not planned and not already included in your IRB approved protocol, IRB approval will be required if you later want to enroll such a subject, or you want an enrolled subject who loses capacity to remain in the study, (unless the subject's loss of capacity is temporary).
- If the potential participant lacks the capacity to consent to research participation, consent must be obtained from their legally authorized representative (LAR).
- The hierarchy for determining who may serve as the LAR at an NIH site is as follows, though for research conducted at non-NIH sites, this may vary due to state law or institutional policy:
- Court-appointed guardian of the person, who is authorized to consent to the research.
- The individual(s) appointed in the patient's Durable Power of Attorney (DPA) for health care.
- If the prospective subject does not have a court-appointed guardian or DPA for health care, and they are capable of understanding the DPA process, even if they lack capacity to consent to research, the prospective subject may execute a DPA for health care.
- If no court-appointed guardian or DPA for health care exists, and the prospective subject is unable to execute a DPA for health care, then the next of kin hierarchy listed below may be used to identify the LAR in the following descending order unless the research is in Category D. See this section of the manual for additional information about Categories of Research with adults who lack capacity to consent to research. (Also see Policy 403 Research Involving Adults Who Lack Decision-making Capacity to Consent to Research Participation which defines these categories of research.) The next of kin hierarchy may not be used to identify an LAR for Category D research per the policy and, in these circumstances, research is not permitted if no court-appointed guardian or DPA for healthcare is available to consent.
- Spouse or domestic partner
- Adult child
- Parent
- Adult sibling, or
- Other relative
- The NIH PI should consult NIH Office of the General Counsel (OGC) if they have questions about the application of this hierarchy. For subjects seen at the CC, see also, MAS 19-1 Determining Legally Authorized Representatives for Adult Patients Who Are Unable to Provide Informed Consent for Clinical Care or Re-Admission (26 March 2020).
Also see this section of the manual for additional information about conducting research with adults who lack the capacity to consent to research.
What other considerations about the consent process exist when doing research with NIH Staff?
- In addition to the usual consent process, NIH staff interested in participating in NIH Research should review the NIH Frequently Asked Questions (FAQs) for Staff Who are Considering Participation in NIH Research which can be found at the end of Policy 404, Research Involving NIH Staff as Subjects in the section labeled Guidance.)
- Prior to enrollment, the NIH investigator must request NIH staff member to review the Leave Policy for NIH Employees Participating in NIH Medical Research Studies (NIH Policy Manual 2300-630-3. (NIH logon required)
- Whenever possible, if staff members wish to participate in research taking place in their own work unit, consent should be obtained by an individual in a non-supervisory relationship with the subject. Also, a consent monitor or other qualified investigator must be present to observe the consent.
- When obtaining consent from an NIH staff member, the investigator should follow the safeguards defined in the protocol.
Also see this section of the manual for additional information about conducting research with NIH staff.
Documenting information in the medical and/or research records
Key points
- Records must be maintained so that they are easily retrievable. Records must be organized and indexed in a manner that permits the access and retrieval of the records in an effective and efficient manner.
- Documentation should be attributable, legible, contemporaneous, original, accurate and complete.
- All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation, and verification.
- The confidentiality of records that could identify participants should be protected.
- Proper documentation allows for source documentation to be available at the time of data abstraction, monitoring visit, audit, or inspection.
- Maintain permanent electronic records (e.g., emails, IMs, text messages, electronic documents, spreadsheets, presentations, images, maps, videos, blogs and other social media tools that generate communications) electronically, and if applicable, in an approved electronic records system.
- Maintain non-email electronic records, in their native format in an organized way on an NIH network drive, SharePoint site or other agency electronic information system.
- It is the investigator's responsibility to complete the NIH annual records management awareness training.
- All NIH Federal records must be managed through the records lifecycle, which includes creation or receipt, maintenance and use and disposition. Additional information about records retention, can be found in NIH Policy Manual 1743-Managining Federal Records.
Record retention
Key points
What are my overall responsibilities as an investigator for maintaining research records and for retaining them after study completion?
- As an NIH investigator, you are responsible for maintaining all study records before, during and after closure of your protocol.
- Maintain a regulatory file with current and accurate records of all study documentation as required by applicable regulatory requirements.
- Research source documents that cannot be placed in CRIS or the electronic documentation system must be maintained in a research record created by the study team.
- All records must be maintained on a secure network location, or if records are in paper form, they should be in a locked cabinet with limited access.
- To determine how long to retain non-FDA regulated study records, use the closure date provided by the IRB. For FDA regulated records, see below. The closure date is the date of the closure letter to the PI and serves as the starting point for this requirement. The letter confirms that the IRB has reviewed the study completion form.
- The determination of how long records will be kept will be based on NIH Manual Chapter 1743-Managing Federal Records.
What are my responsibilities for maintaining records when my protocol is FDA regulated?
For information related to record keeping for FDA regulated research, please see the relevant section under FDA Regulated Research later in this manual.
Data and Safety Monitoring
Key points
- Follow the Data and Safety Monitoring Plan (DSMP) as described in your protocol.
- Provide reports from the data safety monitoring entity described in your DSMP to the IRB at the time of continuing review or sooner based on report findings and proposed recommendation(s).
What are my responsibilities as a PI regarding the Data and Safety Monitoring Plan (DSMP) described in my non-exempt human subject research protocol?
- Confirm that your DSMP as outlined in the IRB-approved protocol is followed.
- See this tip sheet that discusses DSMPs and various data and safety monitoring entities.
- Your DSMP may include a data safety monitoring entity for example:
- Data and Safety Monitoring Board (DSMB)
- Independent Safety Monitor (ISM)
- Safety Monitoring Committee (SMC)
- Verify that required information is promptly provided to the data and safety monitoring entity.
- It is your responsibility to notify the data and safety monitoring entity when there are modifications to your protocol that may affect the DSMP.
- Promptly address and respond to the data and safety monitoring entity recommendations. Your responses may include addressing recommendations, or requests for corrective action plans.
- Provide reports from the data safety monitoring entity (DSME) described in your DSMP to the IRB at the time of continuing review or sooner based on the complexity of the report findings and proposed recommendation(s). The DSME makes recommendations whether a study should or should not continue or should be paused based on its review. The NIH IRB is responsible for ensuring that any proposed changes are implemented for overall safety of the protocol and may take action based on recommendations from the DSME. If a DSME report recommends the study be paused or halted, this must be promptly reported to the IRB consistent with Policy 801, Reporting Research Events.
Suspension or termination of research activities by the IRB
Key points
- The IRB has the authority to suspend or terminate its approval of research, and investigators must comply with IRB requirements.
- In such cases, investigators must coordinate with the IRB to plan for rights, safety, and welfare of enrolled subjects during the period of suspension or for orderly termination of the protocol as directed by the IRB.
- Investigators must obtain IRB approval for any materials that will be used to inform subjects about study suspension or termination.
What does it mean if the IRB suspends or terminates my protocol?
- The convened IRB has the authority to suspend or terminate IRB approval of research that is not being conducted in accordance with the IRB's requirements, federal regulation, NIH policy or that has been associated with serious events, serious problems, or unexpected serious harm. This includes the authority of the Chair to take immediate action to suspend a research protocol to protect research subjects from serious risk of harm, and in this case, the action is reviewed at the next convened IRB meeting.
- The IRB will provide you with its reason for the suspension or termination of your research activities.
- Suspension is a directive by the convened IRB (or the Chair as noted above) to temporarily stop some or all previously IRB-approved research activities (or future enrollment) conducted under an IRB-approved research study. You must obtain IRB approval before the research may resume.
- Termination is a directive by the convened IRB to permanently stop all activities in a previously NIH IRB-approved research study. When IRB approval has been terminated, the study is closed, and a study closure submission form should be completed in the electronic IRB system.
- When a study is suspended or terminated by the IRB:
- You must comply with IRB requirements
- You must inform the sponsor.
- You must coordinate with the IRB to provide a plan for subjects.
- When the research has been suspended, if subjects are enrolled on the research, you must provide a plan to the IRB for its consideration that takes into account the continued rights, safety and welfare of enrolled subjects during the period of suspension.
- If the research has been terminated by the IRB, work with the IRB to create a plan for orderly termination of the protocol, and obtain IRB approval for any materials you will use to inform subjects about the study termination.
- You may be required by the IRB to submit a report in the electronic IRB system detailing events related to the suspension or termination if not already provided to the IRB.
- Suspensions and terminations of IRB approval are reported to OHRP and, if applicable, the FDA.
Administrative hold or early study closures by other entities
Key points
- Entities other than the IRB may request that a protocol be put on hold or closed early for specific reasons.
- A PI may place an administrative hold on their protocol to temporarily halt some or all research activities.
What circumstances may warrant early closure of a research study (e.g., by the PI, the DSMB or the Sponsor)?
Circumstances that may warrant termination include, but are not limited to:
- Determination of unexpected, significant, or unacceptable risk to participants
- Demonstration of efficacy that would warrant stopping
- Insufficient compliance with protocol requirements
- Data that are not sufficiently complete and/or evaluable
- Determination that the primary endpoint has been met
- Determination of futility
What happens if my research study is temporarily halted or prematurely terminated by an entity other than the IRB?
- If the study is temporarily halted or prematurely closed (or if FDA directs that it be put on hold), promptly inform the IRB, and sponsor and provide the reason(s).
- An administrative hold is a voluntary action taken by the PI to temporarily halt some or all research activities. This may result from decisions made by the study sponsor, IC or NIH leadership, or any regulatory agencies.
- During the period of the administrative hold, the protocol is still subject to continuing review, and you must comply with event reporting requirements should relevant new information become available during the hold.
- Administrative holds are not considered suspension or termination of IRB approval and do not require reporting to OHRP or, if applicable, the FDA.
- When a study is temporarily halted or prematurely closed, coordinate with the IRB to create a plan to promptly inform participants. Materials used to notify participants (e.g., letters, verbal scripts) must be prospectively approved by the IRB.
- Ensure there is appropriate therapy and follow-up for participants.
- If the reason for temporary halt or premature closure could be relevant for former study participants, the IRB will determine whether former participants should also be notified.
If my non-exempt human subjects research protocol is prematurely closed, what are the requirements for submission of study closure documents?
When the protocol is being prematurely closed, submit the required form via the NIH electronic IRB system and submit any other documents or information that the Office of IRB Operations (IRBO) requests.
Maintaining your Regulatory Binder
Key points
- A regulatory binder organizes essential documents to provide easy access to essential documents both for the study team as well as the sponsor or monitor.
- Essential documents are those documents which permit evaluation of the study conduct and the quality of the data produced.
- It is highly recommended that all trials have a regulatory binder, regardless of sponsorship.
- Filing essential documents in a timely manner can greatly assist in the successful management of a trial by the investigator, sponsor and monitor. These documents are also the ones which are usually audited by the sponsor's independent auditor and inspected by the regulatory authorities (e.g., FDA) as part of the process to confirm the validity of trial conduct and integrity of study data.
What is a Regulatory Binder?
- A regulatory binder contains all study-specific information and regulatory documentation.
- It organizes essential documents, provides easy access to essential documents by the trial monitor, auditor, IRB, or regulatory authorities (e.g., Office for Human Research Protections (OHRP), Food and Drug Administration (FDA)) for review/audit purposes, and allows research team members to reference information.
A regulatory binder is also referred to as a study binder, investigator binder, administrative binder, regulatory file, or investigator's study files.
- For additional information about regulatory binders and management of research documentation, see Investigator Seminar Series session Documentation and Document Management in Clinical Research-Slides for Downloading and Video
What forms should my Regulatory Binder contain?
Depending on the type of research, your protocol may or may not contain some of these documents, especially if your research protocol is not FDA regulated. Examples of forms to which links are provided in this section are from the Clinical Research Operations site of the NCI Center for Cancer Research.
- Investigator/research staff qualification documentation
- Current curricula vitae (CVs) are collected to demonstrate qualifications of all investigators, associate investigators, and study coordinators.
- Updated copies must be signed and dated.
- Expired CVs should be retained to validate qualification for the entire duration of the study.
- Licenses should be collected, as applicable. Do not remove expired licenses.
- Current curricula vitae (CVs) are collected to demonstrate qualifications of all investigators, associate investigators, and study coordinators.
- Training Records/Certificates/In-services
- If you are the Principal Investigator, you must ensure that there is adequate training for all staff participating in the conduct of the study.
- Retain evidence of training such as:
- Copies of investigators' human subjects research training certificates
- As applicable, additional training certifications for investigators and study staff
- List of attendees/sign-in sheets for relevant study related in-services conducted for investigators and study staff
- IRB documents
- Initial protocol and ALL modifications with documented IRB and Sponsor approval
- All approved versions of the consent form
- All continuing review approvals from the IRB
- Study completion/termination report
- FDA Documents (if applicable)
- Form 1572
- FDA document history log: Track all correspondence submitted to/received from the FDA (e.g., annual IND reports)
- IND Safety Reports and Unanticipated Adverse Device Effect (UADE) Safety Reports
- Investigator's Brochure (IB) or Package Insert
- Include the package insert (in the case of an approved drug) or Investigator Brochure (IB) if one exists.
- If the package insert or IB is amended during the trial or is updated, it should be included in the Regulatory Binder and provided to the IRB in real time.
- Adverse Event (AE) Tracking Log
- This is a log of the adverse events that occur on the protocol.
- This is a log of the adverse events that occur on the protocol.
- Recruitment advertisement/letters
- These materials should have documented IRB approval.
- These materials should have documented IRB approval.
- Sponsor Correspondence
- Telephone logs, meeting minutes and/or relevant email and written correspondence to/from the sponsor
- SAE reports, as applicable
- Site visit reports
- Laboratory Certification and Laboratory Normal/Reference Ranges
- All copies of clinical laboratory improvement amendments (CLIA) certifications for all lab tests, the results of which will be used for purpose of the study
- Copy of normal ranges for all labs/tests included in protocol
- If using results for a specific participant as the reference ranges, blacken out all participant specific identifiers, copy and then place in the binder.
- Specimen Tracking Log, as applicable
- A log of research specimens that includes type of specimen, purpose of storage, location of storage and link to participant ID number. If applicable, the log should be modified to track if consent for future use was obtained, declined, or withdrawn.
- A log of research specimens that includes type of specimen, purpose of storage, location of storage and link to participant ID number. If applicable, the log should be modified to track if consent for future use was obtained, declined, or withdrawn.
- Screening/Enrollment Log
A screening log is a log without identifying information used to track subjects who were screened for the study including screen failures and those enrolled in the study. A screening/enrollment log should include:- All participants screened
- Participants screened who declined further participation (include reason for declining)
- Participants screened who were deemed ineligible for study participation
- Participants screened who did not continue on to enrollment and study intervention or treatment (include reason i.e., death before enrollment etc.)
AND - Track screen failures: Participants who consent to participate in the study, who do not meet one or more criteria required for participation in the trial during the screening procedures, are considered screen failures
- Indicate in the protocol how screen failures will be handled in the trial, including conditions and criteria upon which re-screening is acceptable, when applicable
- Rescreened participants should be assigned the same participant number as for the initial screening.
- A sample screening/enrollment log can be accessed here.
- Site Visit Log
- Log in which monitors will document their visits
- Site staff initials/verifies that a monitor was present on specific dates
- Visit correspondence, such as confirmation or follow up letter
- For consecutive days, each day is entered separately. A sample Site Visit log can be accessed here.
- Delegation of Responsibility Log
- As the investigator, this is where you document what tasks are delegated to members of your research team based on each individual's competencies.
- This log should be kept up to date as new study personnel are added and/or study roles change.
- This link contains a sample delegation of tasks/authority log.
- Protocol Deviation (PD) Log
- This is a log to track all deviations that occur over the life the protocol. A sample PD log can be accessed here.
- This is a log to track all deviations that occur over the life the protocol. A sample PD log can be accessed here.
- Pharmaceutical Information, as applicable (In some cases, this information and documents will be kept by the Pharmacy.)
- Drug accountability including shipping and dispensing records
- Sample of labels attached to investigational product containers
- Decoding/breaking the blind procedures (if not detailed in protocol)
- Data Safety and Monitoring Documents
- Study reports, recommendations and minutes generated from meetings of the data and safety monitoring entity
- Study reports, recommendations and minutes generated from meetings of the data and safety monitoring entity
- Additional contents of your Regulatory Binder may include the following:
- Blank set of Case Report Forms
- Record of retained tissue or fluid specimens
- Notes to File
Who is responsible for maintaining the Regulatory Binder?
- The Principal Investigator is ultimately responsible for maintenance of regulatory files. However, this task can be delegated to other members of your study team.
What guidelines should I follow for maintaining my Regulatory Binder?
- Make sure confidentiality of the subjects is maintained.
- Redact names of subjects and use subject numbers in reports (e.g., expedited event reporting to the IRB).
- Binder contents/organization need to be easily understood by someone who is not familiar with the study.
- Keep binders in a secure location (e.g., locked up).
- File documents in reverse chronological order.
- Do not use binders to hold irrelevant papers (i.e., post-it notes to yourself).
- If you are using a thumb drive during monitoring visits, it must be encrypted.
Subject complaints and communicating with subjects
Key points
- Investigators should communicate with the subject who is expressing the complaint (the "complainant") respectfully. See Policy 104, Managing Research-related Complaints from Subjects for more information.
- Investigators should report all complaints to the NIH PI, or IC leadership for response or referral of the matter, as appropriate.
- If the complaint represents a reportable event as described in Policy 3014-801, Reporting Research Events, the PI will comply with reporting requirements described in the policy.
- Whether written or verbal, investigators should document the complaint (e.g., in the research record, CRIS, or other tracking system).
- The PI should report complaints that remain unresolved to the OHSRP office of Compliance and Training, and they also should report such complaints to the IRB at the time of Continuing Review.
- Complaints received by the Clinical Center Patient Representative that relate to human subjects research are usually referred to OHSRP.
- Investigators should understand what mechanisms permitted by NIH can be used to electronically communicate with study participants, see below for more information.
How are subjects informed about who to contact if they have a research related complaint?
- Designated contacts are identified in the consent form and include the PI and any other study investigator specifically listed on the form.
- Additionally, if subjects have concerns regarding their research participation, they can contact the IRB Office as listed in the consent form. Complainants may also bring their concerns to OHSRP.
- Additionally, subjects seen at the Clinical Center can contact the NIH CC Patient Representative.
- ICs who have investigators who conduct human subjects research at other locations must also have specific individuals or offices external to the research team within the IC who subjects can contact about complaints. Examples include the NIH IRB Office and the Clinical Directors Office.
What should I do if I get a complaint from a subject participating in a study for which I am an investigator?
- Whether written or verbal, you should document the complaint (e.g., in the research record, CRIS, the office tracking system) consistent with federal law, regulation, and policy, including NIH policy. If you have questions, contact your CD.
- Communicate respectfully with the complainant.
- Address complaints to the extent possible within your ability, scope, and authority, as soon as feasible.
- You should also report the complaint to the study PI or IC leadership for response or referral of the matter, as appropriate. If OHSRP receives the complaint initially, it will inform the PI.
- When reporting subject complaints to the PI, staff should honor a subject's request for anonymity to the extent possible. (See the questions that follow for additional instructions.)
- The PI or an AI who is an NIH federal employee will address the complaint as soon as feasible, and/or refer the matter to other NIH or IC offices, as appropriate
- If you are the PI, when you become aware of the complaint, provide appropriate support, management, oversight, and assistance regarding complaints reported to study team members.
What if the subject tells me they want to keep the complaint confidential?
You can inform the subject (or delegate to another the responsibility to do so), when pertinent, that:
- The name of the complainant(s) will be kept confidential to the extent possible, and that complete confidentiality cannot be assured.
- The corresponding subject's identity may be revealed during the investigation but held confidential to the greatest extent possible.
- Anonymous complaints will be accepted. However, anonymity may hinder both the investigation of the complaint, as well as inhibit the ability to provide responses to the complainant.
- The issue will be considered by those receiving the complaint as soon as feasible, but it may be referred to other parties, as appropriate.
- Results of a subsequent inquiry or an investigation, if any, may be provided to the non-anonymous complainant, consistent with the Privacy Act of 1974, federal laws, regulations, and policy, including NIH policy. In some instances, for example, the complainant may be told that appropriate measures have been taken, or that the matter is being investigated and no further information will be forthcoming.
If I am the PI, to whom do I report the subject complaint?
- If the event relates to a possible unanticipated problem, possible noncompliance (including major protocol deviations), it should be reported in the electronic IRB system using the appropriate form as per Policy 3014-801, Reporting Research Events.
- If the complaint remains unresolved, the PI should notify the OHSRP office of Compliance and Training since their staff is available to assist NIH PIs in handling and responding to complaints.
- If NIH is the Reviewing IRB, unresolved complaints should be reported at the time of Continuing Review.
- If a non-NIH IRB is the Reviewing IRB, complaints should be reported consistent with the terms of the reliance agreement.
In addition to the notifications above, what do I do if I think that the subject's complaint may result in a public relations concern?
- If you are not the PI, you should notify the PI who should contact their IC's Communication Director.
- For publicity-likely complaints that involve care inside the Clinical Center, the PI should notify both their IC Communications Director and the Clinical Center Communications Director.
What mechanisms can I use to electronically communicate with study participants?
- For the most current information about options for secure communication with participants, see the Health Information Management Department Handbook (available only via Clinical Center Intranet).
- Medical Secure Email is a web-based system that allows NIH staff to send messages with attachments to participants' email addresses. Once a message is sent to the approved participant email address, they will receive a notification that a secure package (message) is available, and the participant then needs to securely log in to the system in order to see the content of the message.
- It is the responsibility of the NIH staff member to verify the approved patient email address in CRIS prior to messaging a participant.
- Texting is not considered secure and should not be utilized for such communications.
- Secure Health Messaging is a different system that allows NIH users to message participants directly in CRIS or via a secure website. The message to the patient is received in their FollowMyHealth® account. Any NIH staff wishing to message a patient must first be identified as a care provider for that participant in CRIS in order for the participant to view and respond to the message.
Overview
Content Tools
ThemeBuilder