
search
attachments
weblink
advanced
Overview
Content Tools
ThemeBuilder

What's New with iRIS - 2020 Q3
The iRIS team continues to streamline and make more efficient the iRIS workflow. Below are some of the improvements made for you in the late spring and summer:
- New -Auto Numbering of IRB Applications
- iRIS was revised to auto create an application’s IRB number in the system upon creation of the form. This eliminated the manual process of assigning a number that was previously taking place.
- Revised-Not Human Subjects Research (NHSR) application
- The NHSR application was revised to be more streamlined and to be created in the same way as other iRIS applications.
- Revised -Ancillary review trigger Qs
- The amendment form was revised to only require completion of relevant sections if only the following were selected:
- Investigator Brochure Submission Form
- KSP Changes Form
- The amendment form was revised to only require completion of relevant sections if only the following were selected:
“Ask the iRIS Trainers” Brownbag lunch session
These recurring monthly ‘Ask the Trainer’ brown bag lunchtime sessions will be held panel-style. Users should come prepared with their general questions for all to learn from. We hope that you join us!
You can find more information, including upcoming schedules, on the iRIS Training page.
OHSRP Newsletter 2020 Q3
Letter from the Director

September 16th was my 2-year anniversary of coming to the NIH. When I reflect on what we have accomplished together during these past 2 years, it is no wonder that sometimes I feel tired….as I am sure you do also. The road has been and still is bumpy, although I think the potholes are less deep and not as frequent. There is no doubt the pandemic has adversely impacted the ability of our office to consolidate and streamline its operations and develop the critical team structures that are needed to function optimally. Despite this, we continue to...
IRBO Update

During this past quarter, the IRBO sent out a number of notifications to the research community through the NIH iRIS Notification System which included important information for investigators and research support staff. We have summarized this information here for your convenience.
What's New in iRIS?

The iRIS team continues to streamline and make more efficient the iRIS workflow. Here are some of the improvements made for you in the late spring and summer...
Compliance & Training Update

During this past quarter of 2020, the OHSRP Education Series included a 2-part presentation by the IRB Office Reliance and Single IRB team (July and August 2020) that is now available in the archived presentations section on the OHSRP website. This link provides access to the slides and videocasts for all past sessions of the OHSRP Education Series. The Reliance and Single IRB team is also hard at work updating their presence on the IRBO website including revisions of existing resources and preparing new tools to assist investigators/study teams to conduct multisite research. These will be available later in 2020 and will include...
Policy Update

Human Research Protection Program (HRPP) policy development is wrapping up. OHSRP began rolling out the policies late this Summer, the remaining policies will be rolled out through the end of 2020. To aid in understanding key information about each policy, OHSRP has also developed supporting tools for the community, an overview/change table and a narrated presentation...

Letter from the Director - 2020 Q3
September 16th was my 2-year anniversary of coming to the NIH. When I reflect on what we have accomplished together during these past 2 years, it is no wonder that sometimes I feel tired….as I am sure you do also. The road has been and still is bumpy, although I think the potholes are less deep and not as frequent. There is no doubt the pandemic has adversely impacted the ability of our office to consolidate and streamline its operations and develop the critical team structures that are needed to function optimally. Despite this, we continue to make progress and are in every way I can think of a totally respectable IRB, staffed by dedicated, committed professionals.
Recently, the leadership of OHSRP came together and developed a vision statement.
We will promote the safe and ethical conduct of human subjects research by
- providing timely, consistent and compliant reviews
- educating our community
- communicating effectively and responsively
- collaborating with stakeholders
and thus, will be recognized as national leaders in human subjects protections.
We think this captures well what we believe it means to be the best IRB. We evaluate every decision we make against this vision to assure it is in alignment.
We have begun the process of investigating whether we should replace our current electronic IRB submission system, iRIS, with another system. Having a user friendly, streamlined IRB submission system aligns well with our vision by facilitating timely, consistent and compliant reviews. Believe me, we understand the impact of even thinking about such a change, and it is not something we are taking lightly. There are several reasons that we are considering this change. First, user satisfaction across all user groups for iRIS is very low. When we surveyed the NIH community, 70%of researchers, 62%of IRB members and 83%of IRB staff disagreed or strongly disagreed with the statement “Overall I am very satisfied with the iRIS system”. Second, the workflow processes within the system are such that they significantly hinder our review process. In addition, the system is designed in a way that makes it very difficult to accurately document important regulatory determinations. For these and several other reasons, we feel it is important to at least explore if there is a system that can better serve all of our needs. Rest assured, we will engage and collaborate with all stakeholders throughout this process. We will be presenting a session via videocast on October 6th to discuss this project in greater detail. Please be sure to tune into this to learn more.
The NIH Intramural Research Program is now a signatory to the SMART IRB Master Reliance Agreement. We are now part of a collaborative of 800 institutions nationwide that have signed the agreement. What this means for our researchers is that when we are participating in multi-site research that uses a single IRB, if our partners are also SMART signatories, we do not need to negotiate a separate reliance agreement. The SMART agreement serves as the reliance agreement. While this does not remove all the barriers to single IRB review, our hope is that it will significantly reduce burden and streamline the process. Our heartfelt thanks to all who worked so hard to get the agreement to a place where NIH could sign.
Our key metrics for the last period are shown below.



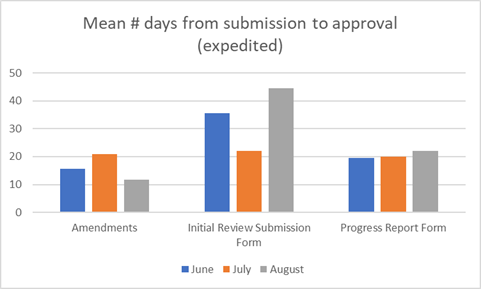

IRBO Update - 2020 Q3
Since our last update, the following new staff have joined the office: Lisa Bingaman- IRB Coordinator, Kelly Pauly-IRB Coordinator, Mollie Fraser -Program Specialist, Kevin Rasmussen-iRIS Trainer. We are excited to have them as part of our team!
During this past quarter, the IRBO sent out a number of notifications to the research community through the NIH iRIS Notification System which included important information for investigators and research support staff. We have summarized this information here for your convenience.
July 7, 2020 - Avoiding Having Your Protocol Lapse
Continuing Review (CR) of research by the IRB provides assurance that research activities remain approvable and that appropriate human subjects protections are in place. Ensuring that continuing review (when required) occurs in a timely manner, is a shared responsibility of both the Principal Investigator and the IRB. When IRB approval for a study lapses, all research activity must cease, except for those activities necessary to ensure the safety of research participants (with permission from the IRB Chair). Lapses in IRB approval are disruptive to the research and place the investigator and institution at regulatory risk as well as risk our status as an AAHRPP-accredited Human Research Protections Program (HRPP).
To facilitate timely continuing review, we ask that study teams submit their CR application to the IRBO 6 weeks ahead of the study expiration date. iRIS will send system generated messages to the study contacts at 75, 60 and 30 days prior to the expiration date to prompt you to submit your CR Application to the IRBO.
If a study team fails to submit the CR application and the study lapses, the IRB will enforce the following policies:
- The IRBO will not review any new studies submitted by the Principal Investigator of the lapsed study until the CR has been submitted or the study has been properly closed.
- If 45 days after study expiration, no CR application or study closure has been submitted, the IRB will administratively close the protocol. Continued research will require submission of a new protocol to the IRB.
August 10, 2020 – Updated Consent and Protocol Templates
- Consent Library: The consent library, found on the OHRSP website, has been greatly expanded to include descriptions and risks of numerous common procedures. Please note that most of this is suggested language. However, the GINA language and language regarding the risks of radiation should not be altered. We have now expanded the radiation risks to include the language for therapeutic radiation and radiation reviewed by the RDRC. This is a living document, and we will continue to add to it, so make sure to periodically check it out.
- Interventional Drug and Device Clinical Trials Protocol Template: This template has been updated with minor updates to make it consistent with new policy and the Natural History Protocol Template.
- NCI CIRB Approved Local Context Consent Templates (English & Spanish): These templates are to be used for studies reviewed by the NCI CIRB which are conducted in the Clinical Center. Please note that you cannot alter the template language in any way.
The Consent Library and the templates referred to above can be found on the “IRB Templates” page of the OHSRP website.
August 31, 2020 & September 18, 2020 - “Tips & Tricks for Submitting to the NIH IRB”
Continuing Reviews (CRs)
- You cannot make any changes to the protocol or consent form(s) at the time of continuing review.
- If you have consented subjects since the last CR, you must submit a redacted consent form(s). Redacted means that you have removed all PII associated with the subject (e.g. his or her signature). The date the subject signed the consent should not be removed.
- If NIH is the coordinating site and other sites have local IRB approval, you must attach copies of the most recent local IRB approvals.
- You must include a high-level summary of any minor and major protocol deviations (PDs), non-compliance, AEs and SAEs that have occurred since the last review, and all unanticipated problems (UPs) that have been reported during the last CR period.
Amendments (AMs) and New Protocols
Protocol
- Remove all NIH personnel aside from the PI from the title page and include these individuals as part of a separate Study Personnel Page.
- Delete the “Investigator Qualifications” section.
- Include the blood volume amounts that will be obtained at each blood draw and the total amount of blood volume obtained over a two-week time period.
- Screening and Natural History protocols: Ensure you have only listed research procedures and removed clinically indicated procedures or have clarified which procedures are being done for research vs. clinical purposes.
- Remove the “Classification of Risk” Section and all IRB determinations of risk, including statements such as, “This study has direct benefit, so only one parent signature is required.”
- If the study involves a non-significant risk (NSR) device, ensure this is described.
- Pregnant Women: If you wish to enroll pregnant women or allow women who become pregnant to remain as part of your protocol, you must revise the protocol to provide a justification that demonstrates that the study objectives cannot be met without including pregnant women. In accordance with 45 CFR 46.204(b), you must make the case that “The risk to the fetus is caused solely by interventions or procedures that hold out the prospect of direct benefit for the woman or the fetus; or, if there is no such prospect of benefit, the risk to the fetus is not greater than minimal and the purpose of the research is the development of important biomedical knowledge which cannot be obtained by any other means”.
- For studies approved before January 21, 2019: If you are conducting screening activities before consenting and obtaining a signature from subjects, include a request for a waiver of informed consent (or a request for a waiver of documentation of informed consent, e.g. if you are using a phone script to consent subjects).
- Remove the use of a witness during the long form and telephone consent processes.
- Remove a request for approval to use the short form consent process.
- Secondary Research protocols: Ensure there is a section requesting a waiver of informed consent along with a justification which addresses all of the criteria in the regulations. (See the “Secondary Research” protocol template on our website).
- If enrolling children, describe the planned assent process and indicate ages of verbal and written assent.
- Update to include safety and event reporting information that is consistent with Policy 801.
Consent Forms (CFs)
- If you are transferring old consents over to the new template, or creating a new consent, form, always use the most current consent template that is posted on the OHSRP website.
- In the most recent consent template version (Revised Common Rule), the language regarding the storage of specimens/data for future use and sharing is now in blue. If you are conducting your study in the United States, you must include this language and may not alter it (except to choose those sections which apply to your protocol). If your study is being conducted internationally, you may alter this language, as appropriate to the locale of the research.
- Remove inclusion/exclusion criteria.
- Update to include the current radiation safety language.
- Include blood volume amounts obtained at each blood draw and the total amount of blood volume obtained over a two week period. The volumes should be labeled in household terms (teaspoons and/or tablespoons); e.g. 5mL=1 teaspoon, 3 teaspoons =1 Tablespoon.
- Address the use of non-significant risk (NSR) devices
- Covered protocols: Include the proper COI language.
- For COVID studies, unless behavioral research where research procedures are limited to questionnaires, include the PREP ACT language.
- Prior to submitting the consent forms to the IRB, keep only the signature blocks that are applicable to your study and delete the blocks which are not applicable.
Responding to Stipulations during Pre-Review
- When responding to stipulations, please keep all changes in tracked form, during the various rounds of submissions to the IRB, i.e. the cumulative changes should be visible to the IRB.
The complete “Tips & Tricks” document can also be found on the “Policies & Guidance” page under “Other Guidance Documents” section of the OHSRP website.

Compliance & Training Update - 2020 Q3
During this past quarter of 2020, the OHSRP Education Series included a 2-part presentation by the IRB Office Reliance and Single IRB team (July and August 2020) that is now available in the archived presentations section on the OHSRP website. This link provides access to the slides and videocasts for all past sessions of the OHSRP Education Series. The Reliance and Single IRB team is also hard at work updating their presence on the IRBO website including revisions of existing resources and preparing new tools to assist investigators/study teams to conduct multisite research. These will be available later in 2020 and will include resources for those relying on an external IRB, such as NIH Institutional Review guidelines and consent templates. For NIH study teams leading multi-site research, resources will include a revised decision tree to help establish if and what type of agreement may be needed e.g., reliance agreement. Additionally, the slides and videocast link for the September session, NIH Policies Related to Enrollment of Pregnant Women, Participants Lacking Capacity to Provide Informed Consent, and Prisoners: Current Status and Recent Updates are also available on that link. The next session on October 6th from 3-4 PM will be available via live NIH videocast at which time Dr. Jonathan Green and Meredith Mullan from OHSRP will present “Transition to a new eIRB [electronic IRB] System: Where we are now, and where we are going.” The videocast and slides will also be available after the session on the OHSRP archived presentations.
As new investigators join NIH, OHSRP has been receiving more questions lately related to human subjects research training requirements, and individuals can refer to Policy 103, Education Program, which spells out the NIH HRPP training requirements based on what type of human subjects research will be conducted. Additional information is available in the guidance document that accompanies the policy. We also have been responding to queries regarding accessing the current site for the required CITI training via the NIH CITI portal. The only portal that NIH investigators should use to access CITI training is the one that links from the Compliance and Training page on the ORSRP website. This is critical because this is the only site that allows CITI training completion records to download automatically into iRIS, the system that the NIH Intramural Research Program uses for IRB document management purposes. These FAQs explain how to access the appropriate NIH CITI portal, how to create an account in CITI via that portal, how to transfer CITI completion records completed at another site into the NIH CITI portal and other questions that will help the user navigate the processes needed to complete and document the required training. The OHSRP Division of Compliance and Training has posted additional educational resources to the OHSRP website including a guide for completing reportable event forms (REF) in iRIS. This provides step by step instructions as well as screenshots and tips to assist investigators in REF submission. We have also added a Reportable Events Flow Diagram to assist investigators in understanding the process of REF submission and review as well as referral to the NIH IRB if the event is a possible unanticipated problem or to the NIH Research Compliance Review Committee if the event constitutes possible serious and/or continuing noncompliance. FAQs related to handling of IND and IDE Safety Reports have also been added, and these clarify when these reports should be submitted to the NIH IRB. Coming soon-we will be posting a topical index that lists the various resources available on the OHSRP Website as well as links to federal guidance documents for each topic.

Policy Update - 2020 Q3
Human Research Protection Program (HRPP) policy development is wrapping up. OHSRP began rolling out the policies late this Summer, the remaining policies will be rolled out through the end of 2020. To aid in understanding key information about each policy, OHSRP has also developed supporting tools for the community, an overview/change table and a narrated presentation.
- The overview/change table is a tool for investigators and study teams that highlights key points of the policy and any changes from the previous SOP(s).
- The narrated slides are a brief overview of the key points of the policy and are aimed at a broader audience, for example IRB members or IRBO staff in addition to investigators. These slide decks also include helpful tips for investigators such as what the IRB expects related to the policy.
OHSRP recognizes that the rollout schedule is aggressive, particularly given the large number of policies. This schedule is due to the fact that the NIH HRPP is about to undergo reaccreditation with AAHRP (see Accreditation Updates below for more information).We understand that this can cause anxiety and raise questions about implementation of changes resulting from implementation of the policies.We hope you saw the email regarding the Guide to Implementation of HRPP policies sent on September 15th. Although the policies have a new format and the policy topics have been reorganized, there are not a lot of new policy requirements. The new policy requirements address the revised Common Rule (45 CFR 46), the reorganization OHSRP and consolidation of the NIH IRBs. In addition, we hope that we have fixed issues from the previous SOPs. Below are helpful tips for implementation of the HRPP policies:
- Each policy has a release date and an implementation date.
- The release date is the date we post the policy on the web and make it available.
- The implementation date is when you should begin following the new policy requirement on your research.
- If a new policy impacts what is written in your protocol or consents, you do not need to immediately revise the protocol or consent(s) to be consistent with the revised policy. In fact, we would prefer that you do not revise your protocol/consent(s) immediately, as this will lead to a surge in amendment submissions which would rapidly overwhelm the IRB office. When to implement these changes in your protocol and consents:
- With your next planned amendment to the IRB. However, even if you do not update the sections with an amendment, the IRB will not stipulate that you update the documents at this time. OR
- In early 2021, when the IRB office will begin requiring that the relevant sections of the protocols/consents be updated.
- However, you should change your actions to implement the new policies on their effective date, even if your protocol or consent(s) is not updated.
- For example, the new policy on enrolling subjects who cannot consent for themselves (Policy 403) changes who can serve as the LAR for research in Category C research, and specifies a new next of kin hierarchy. You should follow the new next of kin hierarchy described in Policy 403 upon the effective date of Policy 403 (September 14, 2020), even if your written protocol does not reflect this practice.
- Lastly, we know that when your protocol/consent does not match your actions, concerns arise about protocol deviations and the need to report them. You do not need to submit a Reportable Event Form (REF) for a protocol deviation during the time period between the implementation of new HRPP policies and early 2021 when IRB will start enforcing compliance with the policies in protocols and consents, or when your protocol is revised to match the new policy requirement, whichever is earlier.
If you have any questions about policy implementation, please reach out to OHSRP at irb@od.nih.gov.
Accreditation Activities
The clock is ticking! On September 15, 2020, OHSRP received the 6-month reminder for the AAHRPP Step 1 Application for re-accreditation. The Step 1 Application is due on March 15, 2021. This Fall, OHSRP will be reaching out to IC AAHRPP Liaisons to kick off data collection for the Step 1 application. Once the Step 1 Application is accepted by AAHRPP, OHSRP will be begin the Step 2 Application process leading up to the site visit. Leading up to the site visit, OHSRP will start educating the members of the NIH HRPP community who have been selected by AAHRPP to be interviewed during the site visit scheduled for January 2022. AAHRPP selects a representative cross-section of the NIH HRPP to be interviewed during the site visit including: NIH and IC leadership, Principal Investigators, research team members, IRB members, IRB staff and OHSRP staff. Keep an eye on the OHSRP Policy page accreditation section for more information.