
What is secondary research?
Secondary research is research with specimens/data initially collected for purposes other than the planned (or primary) research. In this case, the specimens/data were collected to answer a different research question or to test a different scientific hypothesis.
Examples of secondary research vs. primary research are included in the presentation given on 1/19/2021, “Secondary Research: Fact, Fiction, Fears and Fantasies” that is available on the Office of Human Subjects Research Protections (OHSRP) website in the Presentation Archive.
FAQs about Secondary Research
Download this section as a PDF or click on the answers below.
There are two circumstances in which secondary research with specimens/data collected from human subjects may not require prospective IRB review.
- All the individuals from which the specimens/data were collected are deceased, AND/OR
- The specimens/data are not identifiable to the research team (see below for further discussion on this).
Subjects are all deceased
If all the individuals from whom the specimens/data were collected are now deceased, the research with these materials does not meet the definition of human subjects research and does not require prospective IRB review. However, if the specimens/data were collected under another research protocol, the terms of the original consent still apply. For example, if the consent form contained any limitations on the future use of the specimens/data, those limitations must be honored. The investigator is responsible for ensuring that any proposed research is consistent with the original consent.
Specimens/data are not identifiable to the research team
Research, with specimens/data which are not identifiable to the research team, is not considered human subjects research and does not require IRB review and approval. For example, the specimens/data have been fully anonymized by removing all identifiers, or they have been coded, and the investigator(s) conducting the research do not have access to the code key and cannot otherwise re-identify the subjects. The term “coded” means that all identifying information has been replaced with a number, letter, symbol, or combination thereof (i.e., the code) and a key to decipher the code exists, enabling linkage of the identifying information to the specimens/data. See the Guidance for Determining Whether Data Constitutes Individually Identifiable Information Under 45 CFR 46 on the OHSRP website. Investigators should consult with OHSRP if they are unsure whether data or biospecimens being used in a specific research project would be considered individually identifiable.
Secondary research is considered human subjects research that requires IRB review when the specimens/data are identifiable to the researchers and were collected for another purpose than the planned research. The following is an example of secondary research:
- An investigator learns of preliminary data from a study that suggests cigarette smoking leads to specific epigenetic changes that increase susceptibility to certain infections. She also has a large number of pre-treatment samples from cigarette smokers that would be ideal to initially test this hypothesis. The specimens were collected under protocols focused on the study of lung cancer. The samples are coded, and the investigator holds the code key with identifiers. Her planned research with the specimens/data is not described in the objectives of the original lung cancer protocols. The planned activity meets the definition of secondary research because the specimens/data were collected under another protocol for a different research purpose.
Note that if the planned research is related to the existing primary, secondary, or exploratory objectives described in the IRB-approved protocol (under which the specimens/data were originally collected), then it is not considered to be secondary research. This is research that should be conducted under the primary protocol (i.e., primary research). If the investigator is unsure whether the protocol should be amended to address the planned activities or whether the current description in the protocol is adequate to allow the new research to move forward, the investigator should contact the IRB. The following is an example of a proposed activity that does not meet the definition of secondary research:
- An investigator has collected samples as part of an IRB approved phase 1 protocol to determine the maximum tolerated dose of new checkpoint inhibitor drug XYZ123 for the treatment of metastatic lung cancer refractory to standard therapies. The protocol includes an exploratory objective to determine changes in immunologic profiles of subjects receiving the drug. After the trial is already complete, the investigator decides to have all the previously stored samples analyzed for T cell subsets. Since this additional analysis meets the exploratory objective of the original protocol, the planned activities would be considered primary research.
If your research involves the use of specimens/data to test a device, please see Question 4.
If your research involves the use of specimens or data from one or more humans to test the safety or effectiveness of an investigational medical device (e.g. AI/machine learning, in vitro diagnostic (IVD), etc.), the study is considered a clinical investigation under the FDA regulations (see 21 CFR 812.3(h)). This research likely requires creation of a protocol, prospective IRB review and approval (21 CFR 56) and a device determination and either IRB consent or waiver of consent (21 CFR 50). This is because under the FDA regulations, a human subject is not defined based on identifiability. For more information, please consult with the IRB.
To determine if your planned activity with specimens/data is secondary research that will require IRB review, ask yourself the following four questions in order. If the answer to all four of these questions is “yes,” then you are performing secondary research that requires review by the IRB.
- Is the planned activity research? Consider the following regulatory definitions of research:
- Research (2018 Common Rule) means a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge. (45 CFR 46.102(l))
- Clinical investigation (FDA Regulations) means any experiment that involves a test article and one or more human subjects[1], and that either must meet the requirements for prior submission to the Food and Drug Administration under section 505(i) or 520(g) of the act, or need not meet the requirements for prior submission to the Food and Drug Administration under these sections of the act, but the results of which are intended to be later submitted to, or held for inspection by, the Food and Drug Administration as part of an application for a research or marketing permit. The term does not include experiments that must meet the provisions of part 58, regarding nonclinical laboratory studies. The terms research, clinical research, clinical study, study, and clinical investigation are deemed to be synonymous for purposes of this part. (21 CFR 56.102(c))
- Does the planned activity meet the definition human subjects research? Consider the following regulatory definitions of human subject:
- Human Subject (2018 Common Rule) means a living individual about whom an investigator (whether professional or student) conducting research:
- Obtains information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens; or
- Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.
- Human Subject (FDA Regulations)means an individual who is or becomes a participant in research, either as a recipient of the test article or as a control. A subject may be either a healthy human or a patient. (21 CFR 50.3(g))
- Subject (FDA regulations related it use of investigational devices) means a human who participates in an investigation, either as an individual on whom or on whose specimen an investigational device is used or as a control. A subject may be in normal health or may have a medical condition or disease. ( 812.3.(p))
- Human Subject (2018 Common Rule) means a living individual about whom an investigator (whether professional or student) conducting research:
3. Are you using specimens/data that were collected for other purposes (either research or non-research) for the planned research?
4. Is the planned research use of these specimens/data unrelated to the aims or objectives of the current IRB-approved protocol, under which they were originally collected?
1. Please note that the FDA considers research involving human specimens and the use of a medical device to be “clinical investigations”, if data from the research will be used to support an IDE (including IVDs), device marketing application, or another submission to the FDA. In this regard, research involving leftover human specimens that are de-identified is included.
Secondary research is subject to the same regulations and reviewed using the same processes as all other human subjects research. However, depending on the specifics of the study, it may be able to be determined to be exempt from IRB review. Refer to Exempt Research on the OHSRP website.
- Exempt: Submit an exempt protocol via the electronic IRB submission system. Exempt protocol templates and instructions for submitting for an exemption are available at the link above.
- Non-exempt: Submit a secondary research protocol via the electronic IRB submission system. Non-exempt research needs to go through either an expedited or full board IRB review process. Secondary research studies are typically considered minimal risk and are usually eligible for review and approval using expedited procedures. The secondary research protocol template is available on the OHSRP website.
Secondary research that meets the definition of human subjects research is subject to the same regulatory requirements as all other human subjects research; therefore, it must undergo IRB review. The ethical underpinning of this expectation is to assure that the proposed use of the specimens/data meets regulatory requirements and does not violate subjects’ rights. When subjects, as part of an IRB approved protocol, provided their specimens/data, it was with an understanding they would be used for a specific named purpose. IRB review of a secondary research protocol is conducted to assure that the new use is not counter to that intent nor likely to introduce new risks, not previously disclosed to the subject or considered by the IRB. The IRB will also ensure that appropriate privacy and confidentiality protections are put in place.
The original protocol title(s) and protocol number(s) from which the specimens/data were collected, when applicable, should be listed in the new protocol. When investigators will be using existing specimens/data collected under other research protocols, the IRB will look for information about how the investigator has access to the materials. Furthermore, the new protocol must include a summary of the consent language in all applicable consent versions (for all the original protocols) as it applies to sharing and use of specimens/data for future research. This information could be provided in a table or in summary form in the protocol or the previous consent document versions could be uploaded as part of an Appendix. This will allow the IRB to review the original consent language to understand what information was conveyed to subjects about the use of their research specimens/data. Any promises made in the consent regarding sharing and the future use of specimens/data must be honored.
Some examples of circumstances that could be considered include:
- If a previous subject(s) opted out of the use of their specimens/data for future research as part of the original consent, the specimens/data should not be used as part of the secondary research.
- If the original consent stated that the specimens/data would not be used for future research or would only be used for a specific type of future research (i.e., other than what is planned for this study), then the specimens/data cannot be used for secondary research without re-consent.
- If using the specimens/data for secondary research appears consistent with the terms of the original consent, then IRB can waive consent for the secondary research.
- If consent was silent on secondary research, then IRB will consider whether the proposed use
is acceptable with a waiver, or alternatively, if re-consent is required. - Please note that under the revised Common Rule (which applies to new protocols approved
on or after January 21, 2019), there are new consent requirements which could affect what is
allowable within a secondary research protocol. Please see FAQ 9. - The IRB will consider if the secondary research would impose new or significantly greater risks
(including privacy risks) not described in the original consent form. - The IRB will also consider if the study population(s) may have known concerns about the proposed secondary use. An example of this would be Native American or Alaskan Native populations who have distinct culture, beliefs and values and have concerns for potential community harms based on past abuses and violations related to clinical research. Secondary research may require additional ethical review by an Indian Health Service or Tribal IRB and permission from a tribal government.
As with all human subjects research, either the specific consent of the subject to participate in the research must be obtained, or the IRB must waive consent. The future use language in consent forms does not contain all the required elements of consent. Typically, the future use language is very broad, so it does not adequately describe the purpose of the planned study. In addition, secondary research is a new research project that the IRB has not previously reviewed, so a determination that the project meets the IRB approval criteria must be made.
Any permission granted for future use is best thought of as a statement of intent by the subject, that allows the IRB to waive consent for the future use of the specimens/data for research that is compatible with the original consent.
Under the revised Common Rule (which applies to the consent forms associated with new protocols approved on or after January 21, 2019), the original consent form must include:
- A statement that clarifies whether or not identifiers might be removed from the data or biospecimens and that, after such removal, the information or biospecimens could be used for future research studies or distributed to another investigator for future research studies without additional informed consent
- A statement that the subject’s biospecimens (even if identifiers are removed) may be used for commercial profit and whether the subject will or will not share in this commercial profit;
- For research involving biospecimens, a statement that the research will (if known) or might include whole genome sequencing (i.e., sequencing of a human germline or somatic specimen with the intent to generate the genome or exome sequence of that specimen), if applicable.
Furthermore, for new studies approved on or after the compliance date, OHSRP advises that if the investigator intends to share coded or identifiable specimens/data for future research, there should be language in the original consent form informing the subject of this. See the current Consent Templates for Use at NIH Sites.
If the primary research protocol has therapeutic intent or a prospect of direct benefit to the subject, then agreement to unspecified future use should be optional. This is to avoid any possible coercion of subjects who wishes to participate in research that has the prospect to benefit them but who otherwise might not wish to have their specimens/data used for future unspecified research. If the primary research protocol does not have any prospect of direct benefit, then agreement to future use does not have to be optional. In other words, the team could remove the ‘yes’ or ‘no’ check boxes, and the language would then communicate that if the subject chooses to participate in the research study, then their specimens and data may be used or shared for future research.
The sharing of specimens/data with other investigators does not require IRB approval per se. However, the new use of the specimens/data for research may require IRB approval. In order to share specimens/data from a protocol, the consent must allow for sharing, or at least not prohibit it. The proposed research use of the shared specimens/data should be consistent with the terms of the consent under which it was collected. Subjects agreed to participate in a specific study, not anyone's study. If the original consent says the specimens/data will never be shared, you must honor the terms of the consent. This means that the specimens/data cannot be shared even if de-identified. If you still would like to share the specimens/data, you would have to re-consent the applicable subjects with a consent document that is transparent about the plan for sharing. See the flow diagram below and FAQ 12 for examples.
In addition, please note that under the revised Common Rule (which applies to new protocols approved on or after January 21, 2019), there are new consent requirements which could limit the type of secondary research that is allowable as a result of the sharing. Please see FAQ 9.
Can I Share Specimens or Data from My IRB-Approved Protocol for Secondary Research*?
Review the consent form associated with the protocol to determine which scenario below is true. There is….
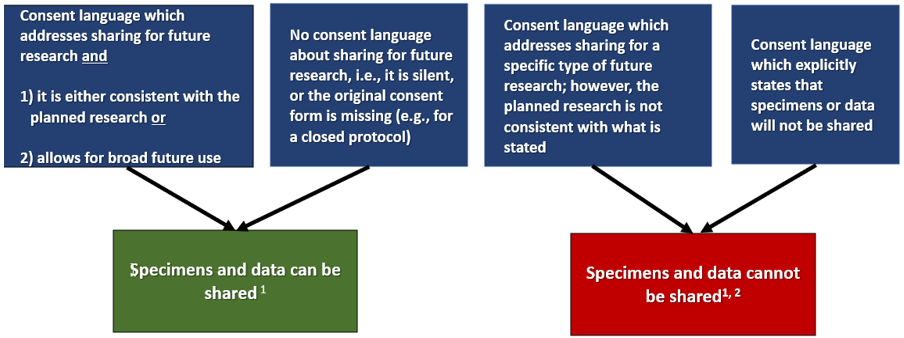
*Secondary research: research use of biospecimens or data for other than the original purpose(s) for which the biospecimens or data were initially collected through interaction or intervention with living individuals
1 If you will receive research results that you can link back to identifiers after sharing specimens or data, the project is considered to be human subjects research. You must submit a secondary research protocol to address the planned research and seek IRB approval prior to initiation of the activities. The protocol must include new consent or a justification of a waiver of consent for the planned research.
2 In some circumstances, it may be appropriate to re-consent the subject to allow their specimens and data to be shared. Consult the IRB if you wish to proceed with sharing.
If the terms of the original consent prohibit sharing, then you should consult with the IRB to determine if there is a path forward. If the IRB provides guidance that sharing might be allowed, they will require you to re-consent subjects prior to any sharing.
The other situation in which you should seek IRB approval is if you are getting identifiable results/data returned to you from your collaborator, and the research is not described in the primary protocol. In this case, you are considered to be conducting new secondary research yourself. In other words, if you are sharing coded specimens (for which you have the code key) with an investigator who does not have the code key, and you are getting individual level data back (not aggregate data), this is human subjects research. The rationale is that you are receiving new information about your subjects that you can link back to identifiers. This activity needs IRB approval not because the specimens are being shared, but because identifiable data is being returned to the NIH research team that will be used for research.
Note that if you are only sharing or collaborating in research involving specimens/data for which the NIH research team has no access to identifiers or the ability to re-identify, this is not considered to be human subjects research. Furthermore, the investigator does not need to submit for a request for determination of "not human subjects research" as was required in the past.
If you wish to share or receive human biospecimens and/or human data outside NIH under a Material Transfer Agreement, a Data Transfer/Use Agreement or a Research Collaborator Agreement, an Investigator Attestation must be completed and provided to the appropriate Tech Transfer contact. Please see NIH Tech Transfer on the OHSRP website for more details and a copy of this document.
Sharing and secondary research with existing specimens/data must comply with the terms of the original informed consent document. Researchers are expected to review all previous versions of the consent form to determine who consented to what. If the original consent form addresses the use and sharing of specimens/data for future research, the new plan for sharing and research should be consistent with the language in the original consent document. If there is language in the original consent form which is contrary to sharing or future research generally or conflicts with the specific sharing and research plan, the investigator cannot proceed. This is true, even if the specimens/data are being used or shared in a de-identified manner, or if the subjects are deceased.
Some examples of prohibitive or restrictive language include:
- “Your specimens will be destroyed at the end of this study.” These means the specimens cannot be stored for future research.
- “Your specimens/data will be used for future research on cancer.” This would restrict any secondary research to only cancer-related research.
- If some subjects opt-out of future research with specimens/data by checking a box in the consent, then the NIH investigator must track this information over time. Failing to check any box is considered the same as not agreeing, i.e., opting out of future research.
- “Your data will never be shared outside of the NIH.” This would restrict the use to only NIH investigators. You would not be able to share even de-identified specimens/data outside of NIH.
- “Your data will never be shared outside of the NIH research team working on the protocol.” This would restrict the use to only the NIH research team members named on the primary protocol.
If the initial consent contained restrictive language and you wish to be able to do future research or share the specimens/data, consult with the IRB. Depending on the type of limitations, the IRB may require you to re-consent the subjects to allow the sharing or research to go ahead. After that point, the specimens/data, of those that provide consent, would now be able to be used.
If the original consent form (for new protocols approved before January 21, 2019) was silent on the topic of sharing and future research, then IRB will consider whether the proposed use is acceptable with a waiver or if re-consent is required.
Per the revised Common Rule (for new protocols approved on or after January 21, 2019), there are new consent requirements which affect what is allowable as part of secondary research. Please see FAQ 9.
Furthermore, for protocols approved on or after the revised Common Rule implementation date, OHSRP advises that if the investigator intends to use coded or identifiable specimens/data for future research, there should be language in the consent form informing the subject of this. See the current Consent Templates for Use at NIH Sites.
Consent to use specimens/data for future research is not sufficient to allow the investigator to move forward with secondary research with identifiable specimens/data. This type of consent language simply allows the investigator to store the materials for future research (or use the materials once completely anonymized (stripped of all identifiers)). If the investigator will conduct new research using existing identifiable specimens/data, generally they are expected to submit a new research protocol and seek IRB approval.
A waiver of consent is when the requirement to obtain informed consent for research is formally waived by the IRB. The waiver applies to the proposed research activity, not to the sharing of specimens/data. The IRB will consider whether the proposed use is consistent with the terms of the original consent and whether there are any new risks. If the conditions described below in the next FAQ are met, then the IRB may grant the waiver. The IRB will not grant a waiver that is counter to the terms of the original consent, nor can the IRB grant a waiver for broad, unspecified future use.
When requesting a waiver for a research protocol being reviewed and approved on or after January 21, 2019, address and provide justification in the protocol for the following specific regulatory criteria:
- The research involves no more than minimal risk to the subjects.
- Example of a justification: The only risk to subjects is a possible breach of confidentiality.
- The research could not practicably be carried out without the requested waiver or alteration (i.e., It is not possible to conduct the research unless consent is not required).
- Example of a justification: The required number of specimens to conduct the research is so large that it would impede scientific validity and introduce bias if only those who were willing to consent were included.
- Example of a justification: Many of the subjects are lost to follow-up, or the research team has not been in contact with them for many years.
- The research could not practicably be carried out without using such information or biospecimens in an identifiable format.
- Example of a justification: The research involves specimens and different types of data (medical records, imaging, lab results) that all must be linked together by a subject identifier to allow analysis.
- The waiver will not adversely affect the rights and welfare of the subjects.
- Example of a justification: Conducting the planned research with existing specimens/data (originally collected under informed consent) will not cause any harm to subjects.
- Whenever appropriate, the subjects or legally authorized representatives will be provided with additional pertinent information after participation.
- Example of a justification: We do not intend to contact subjects to share the results of our research.
- Example of a justification: The results of this research will not generate new clinically actionable findings.
No. Identifiable specimens/data can be stored even after the primary protocol is closed; you do not have to discard or de-identify them. The primary protocol must only remain open if you are using the specimens/data for research purposes described in the protocol. The specimens/data can be stored unused until there is approval of a secondary research protocol. However, although it is permissible to store them, you cannot access or use the identifiable specimens/data for research without an IRB-approved protocol in place.
If the proposed research is directly related to the primary protocol’s aims/objectives, then you can consider adding the new research to the primary protocol. However, if the new project is unrelated to the primary protocol’s aims/objectives, it would be considered new research and needs to be submitted as its own protocol. A protocol needs to be cohesive, and disconnected ideas and experiments are not considered approvable research.
The OHSRP website has templates to guide you in writing your secondary research protocol.
- There is a secondary research protocol template under the Observational Research Protocol Templates section on the OHSRP website. This template should be used for non-exempt research.
- If you think your secondary research may be exempt, refer to the protocol template for retrospective data or biospecimen review on the OHSRP website.
- The protocol template includes information about what types of secondary projects are eligible for an exemption. The webpage also includes instructions that will help guide you through requesting an exemption in the electronic system.
A repository is an organized system to collect, maintain and store specimens/data, most often for future research use. The materials may be prospectively collected from humans for inclusion in a repository, or the repository may house existing materials originally collected for other purposes, including non-research purposes. Repositories can also be referred to as registries, data banks, databases, or biobanks.
Repositories that are designed to prospectively collect specimens/data from humans for research purposes and/or to maintain and distribute specimens/data (that are linked to identifiers) to researchers must have IRB approval and oversight. Accordingly, a repository protocol should first be submitted for IRB review. The repository protocol itself generally does not describe the details of the secondary research. Any secondary research involving the identifiable specimens/data from the repository would require submission of a separate protocol to the IRB. Another option would be for the investigator conducting the secondary research to receive all of the specimens and data in an anonymized or coded and linked format, with no access to the code key. In this case, the investigator(s) overseeing the repository protocol would be acting as an “honest broker” and either permanently anonymize the specimens and data before sharing them or coding them and maintaining the code key, so that only they are in a position to re-link to the original identifiers.
There is a repository protocol template on the OHSRP website under the Observational Research Protocol Templates.
Overview
Content Tools
ThemeBuilder